Extended
X-ray Absorption Fine Structure (EXAFS)
(H. Wende, C.
Sorg, K. Baberschke)
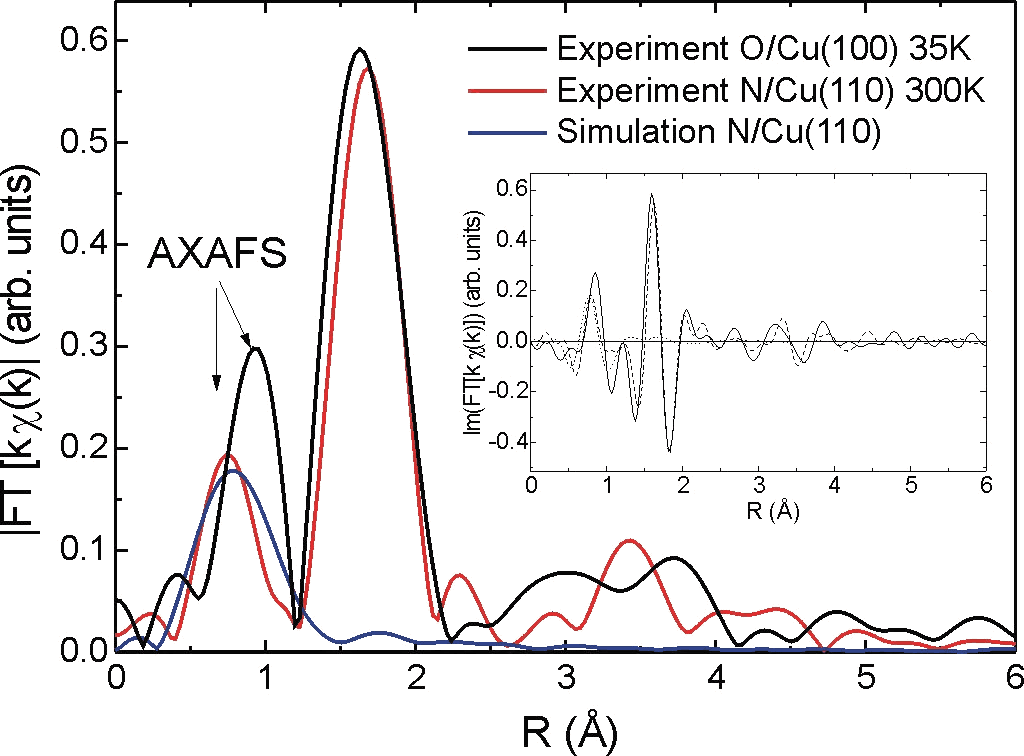
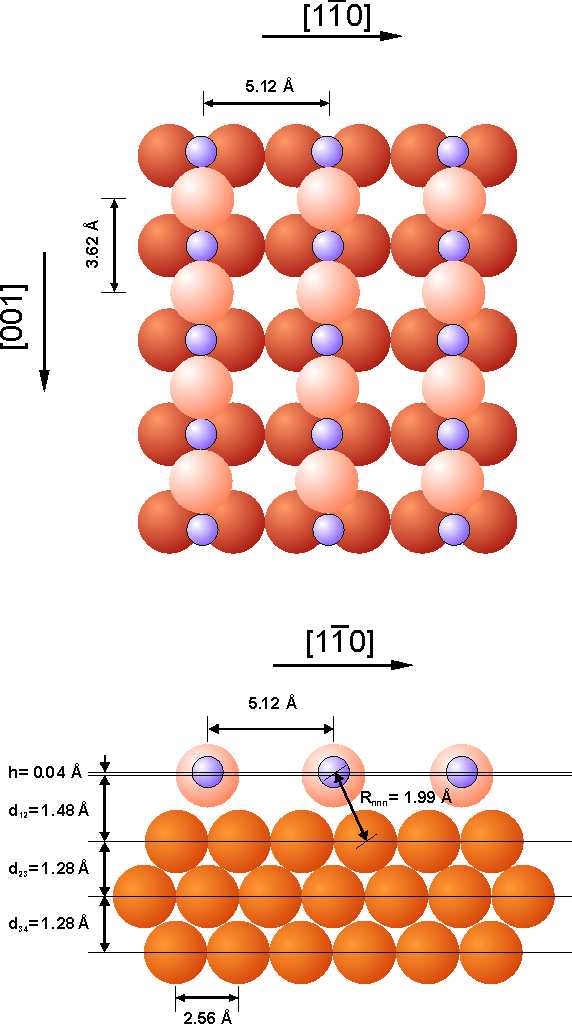
- Surface EXAFS and Near-edge EXAFS from adsorbed atoms and molecules on metallic surfaces
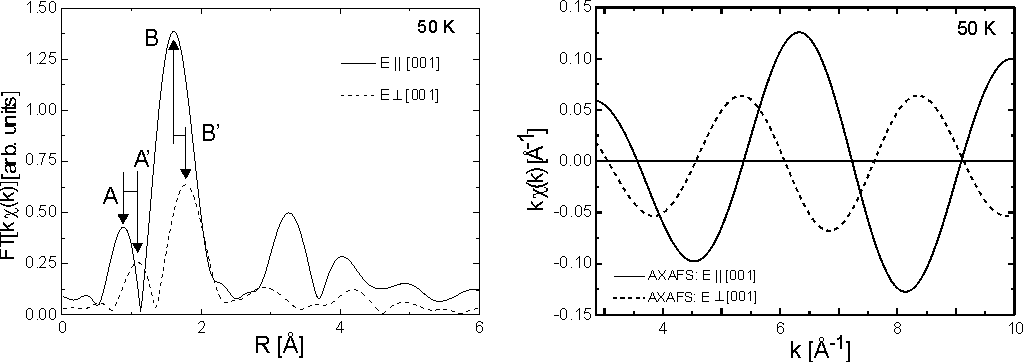
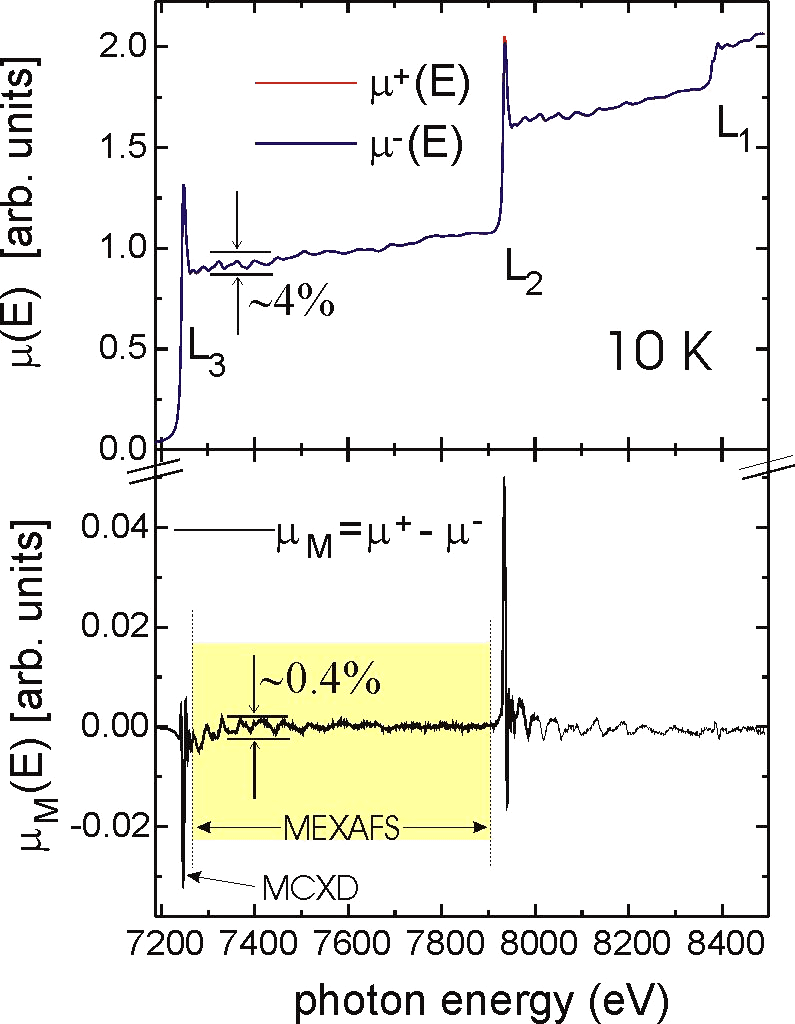
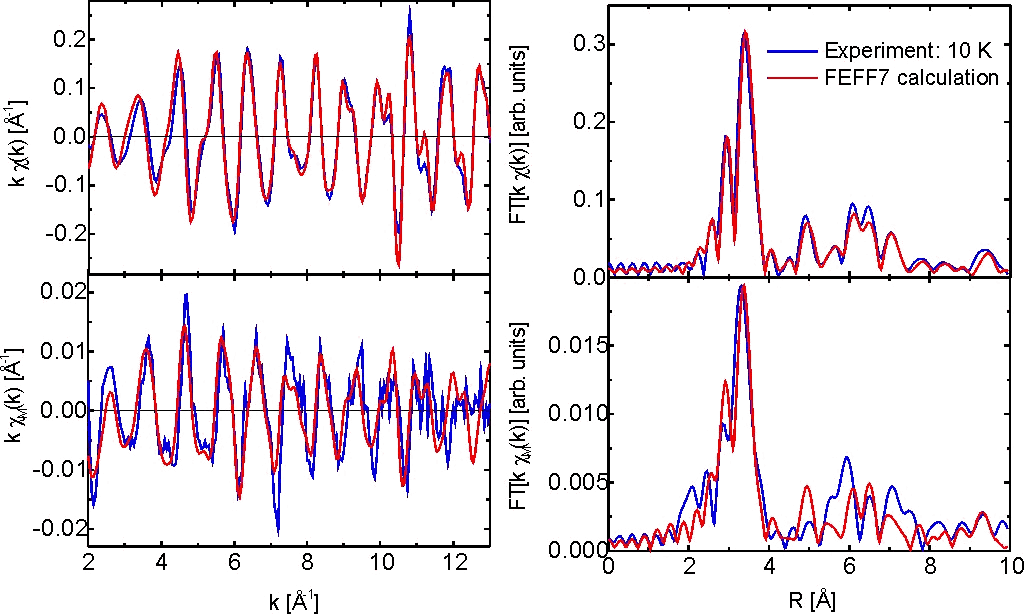
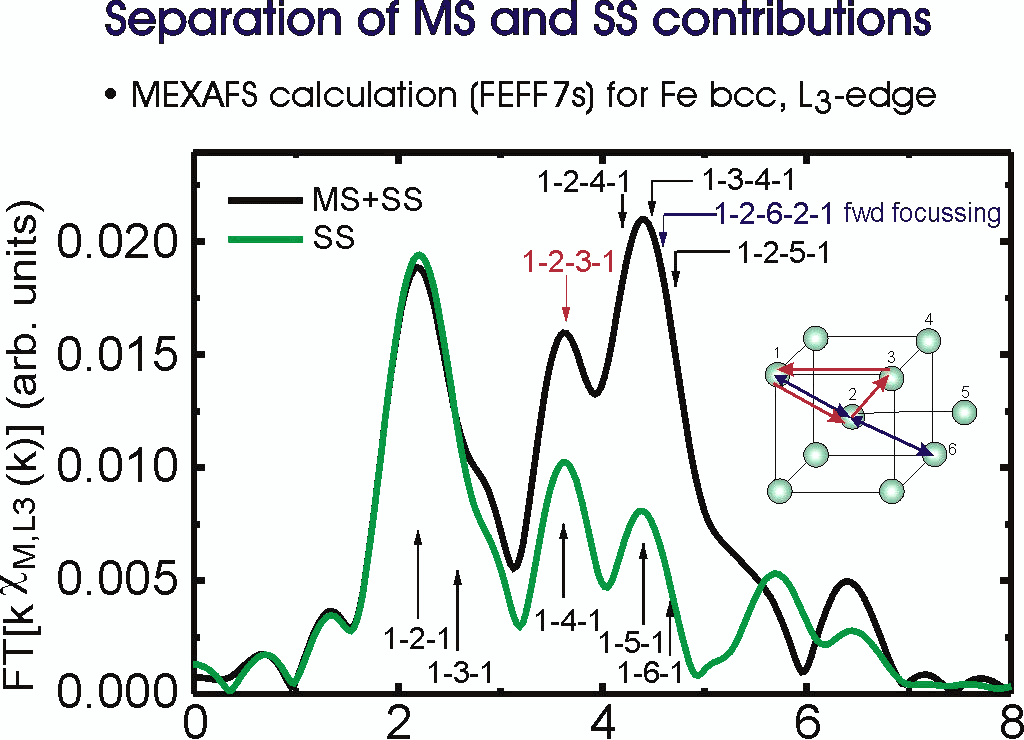
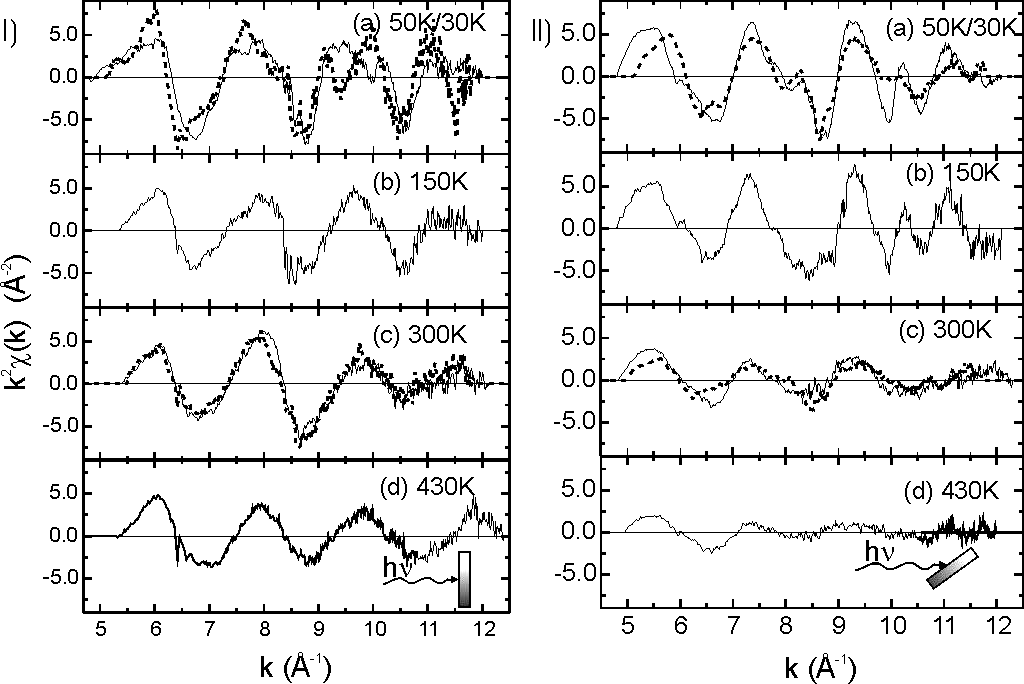 Fig. 1
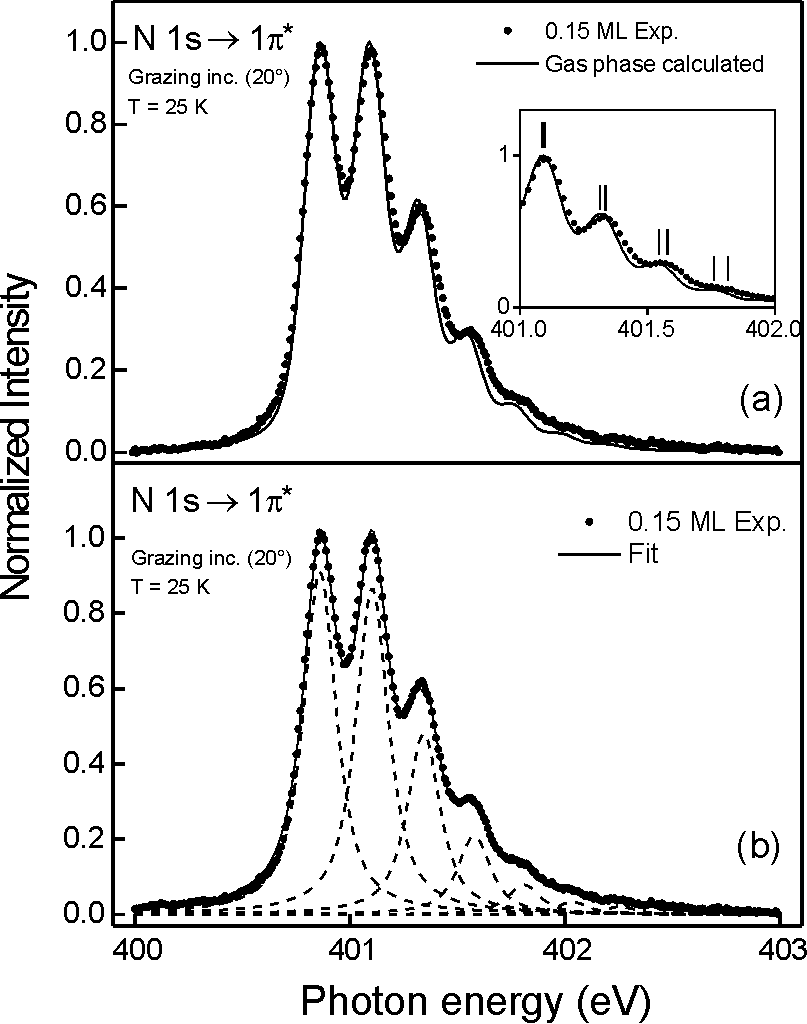
Fig. 1In the second example [Ref. 232] we deal with the vibrational fine structure in the N 1s pi* resonance of the N2 molecule physisorbed on the Cu(100) surface. In Fig. 2(a) we plot the vibrationally resolved photoabsorption spectrum in the N 1s pi* resonance region of 0.15 ML N2 physisorbed at T=25 K on theCu(100) surface (dots) and gas-phase spectrum calculated using the vibrational constants, FWHM, relative intensities for each vibrational peak (by C.T.Chen et al., Phys. Rev. A40, 6737 (1989)) and assuming a Gauss broadening (spectral resolution) as in our measurements (solid line). The inset shows the discrepancy in the peak position for higher vibrational levels. Fig.2(b) depicts the fitted spectrum. The dots and the solid line represent the data and the fit, respectively. The dashed curves are the individual Voigt peaks. Our results for the low N2 coverage show that noangular dependence in the spectroscopic parameters are present indicating, consequently, an isotropic interaction of the pig orbitals with the substrate. On the other hand, for the multilayer absorption there is a preferential orientation.
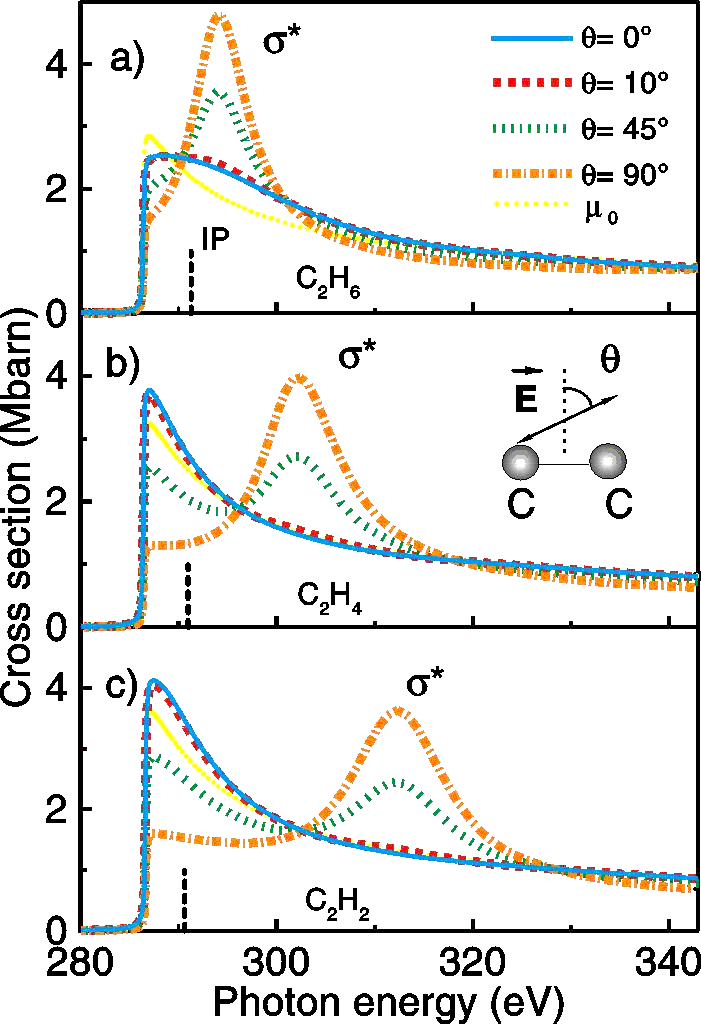
In a third example [Refs. 225, 240] we study the shape resonances of
oriented molecules by ab initio theory and NEXAFS experiment at the C K-edge
of hydrocarbon molecules. We demonstrate that the sigma-shape resonance originates
from the multiple scattering of the photoelectron within the molecule. The
angular dependence of the electric dipole transition in the calculations,
as well as the angular dependent experiments for the oriented molecules
give a good opportunity to compare both. The resonance can be assigned
to a sigma* shape resonance. The multiple scattering formalism and the
experiment agree well and thereby support the existence of such features
in the spectra. The results of the calculated C 1s NEXAFS spectra for different
orientations of the E-vector of the bottom beam with respect to the intramolecular
C-C bond axis that illustrate the above ideas are presented in Fig. 3. The
vertical lines represent the calculated IP values. The atomic background
µ0(E) is indicated by the yellow dotted line. For all three
hydrocarbons a broad feature in the continuum regime is visible, which
shows an asymmetry with a tail on the high energy side in full agreement
with general considerations in textbook scattering theory. The angular dependence
of this structure is in correspondence to predictions for the angular dependence
of sigma* shape resonance.